Device Classification & Strategic Road mapping
We help you determine the correct device classification in line with MDR Annex VIII and select the most appropriate conformity assessment route. Our experts assess whether clinical investigations are required based on your device’s classification and intended claims, then develop a customized regulatory roadmap that guides your team efficiently toward CE marking.
EU MDR Gap Assessment & QMS Alignment
Our gap assessments benchmark your current documentation and systems against the MDR’s General Safety and Performance Requirements (GSPRs), identifying areas of non-compliance. We align your ISO 13485 quality system with MDR-specific processes, recommend updates to standard operating procedures and forms, and ensure your QMS integrates essential elements like PMS, PMCF, and vigilance procedures.
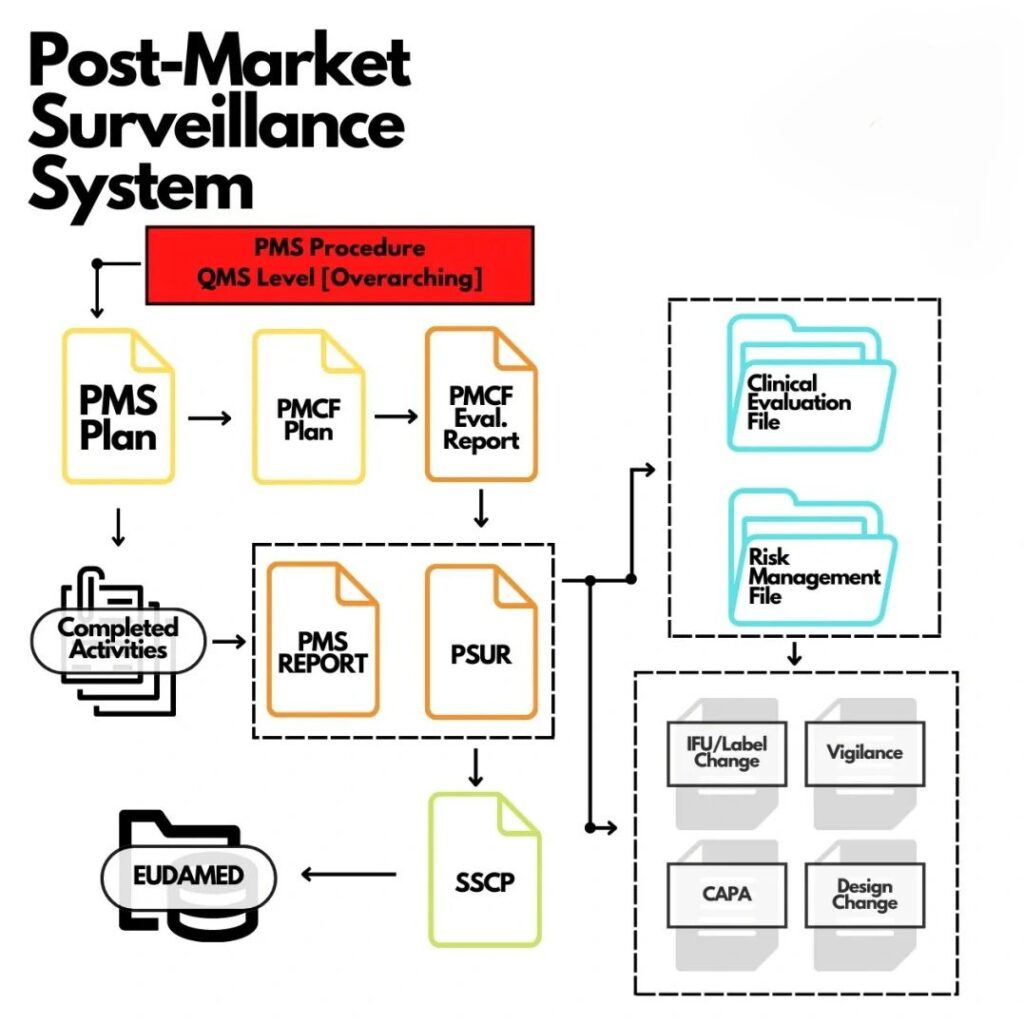
Post-Market Surveillance (PMS) & Vigilance
We develop tailored PMS plans and Periodic Safety Update Reports (PSURs) based on your device’s risk class. Our team designs and validates Field Safety Corrective Action (FSCA) and vigilance procedures, supports incident investigation and trend analysis, and ensures PMS data is appropriately integrated into your clinical evaluation reports and QMS. We also help you harmonize post-market reporting across multiple regulatory markets.
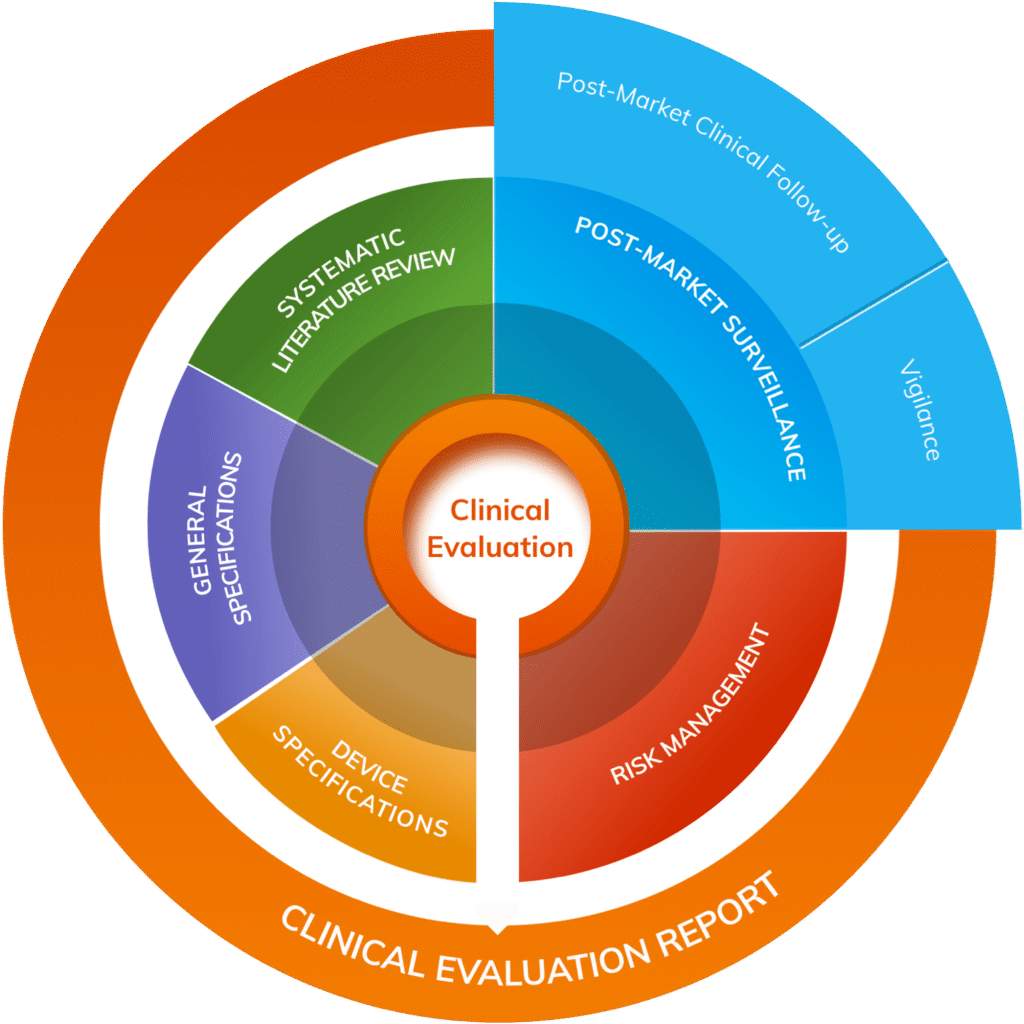
Clinical Evaluation Reports (CERs)
Our clinical team supports the development of Clinical Evaluation Plans (CEPs) and Clinical Evaluation Reports (CERs), whether you’re starting from scratch or validating existing documentation. We assist with literature reviews, equivalence justifications, and clinical data analysis, and can help design and manage PMCF activities, ensuring your clinical evidence remains robust and MDR-compliant.
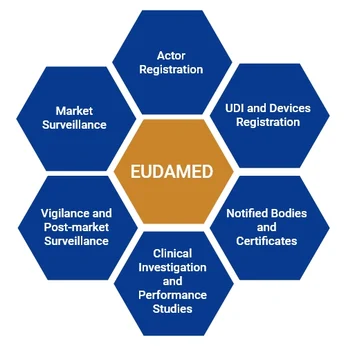
UDI & EUDAMED Compliance
We support you in establishing a UDI system from the ground up, including assignment and management of UDI-DIs and UDI-PIs in accordance with MDR requirements. We assist with EUDAMED registration, generation of Basic UDI-DIs, and creation of UDI traceability documentation, while ensuring your data submission processes remain aligned with evolving regulatory expectations
Technical Documentation Development
We build or update technical files and design dossiers that meet MDR and Notified Body expectations. This includes developing comprehensive labeling, Instructions for Use (IFU/eIFU), device descriptions, and safety and performance documentation. We ensure all technical documentation is well-structured, traceable, and fully ready for regulatory review.


Notified Body Preparation & Support
We guide you through the Notified Body process by helping prepare a clear, compliant submission package and assisting with Notified Body selection and contract negotiation. During the review process, we respond to questions, deficiency letters, and CAPA requests. We also offer audit readiness training, conduct mock audits, and provide expert support during external audits.